実世界には多種多様な化学物質が存在するが、そうした化学物質の物性値を知るには、実際に合成して物性値を測定するか、時間のかかる理論計算をする必要があった。それに代わり、機械学習を用いて分子構造と物性値の関係を学習させる方法が研究されている。
この問題に対して、グラフニューラルネットワークを用いて、化合物の物性値を予測する手法を提案した。例えば原子化エネルギーについては100分の1秒の時間で誤差0.01
eV(電子ボルト)以下の精度で予測できた。これは、理論計算と同程度の精度を、理論計算の1万倍以上の速さで実現したことになる。
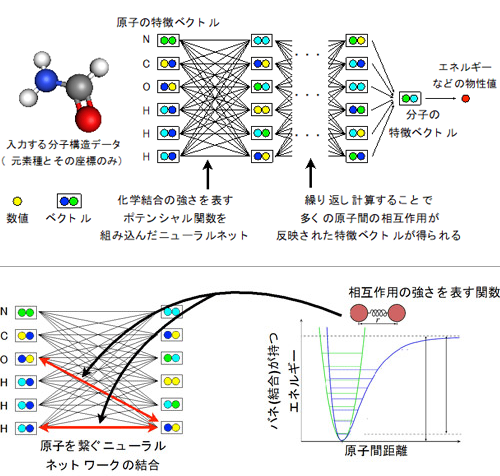
物理化学の知識に基づいて、分子中の原子間に、化学結合などの相互作用の「強さの変化」を「バネの伸び縮み」で表すような関数(ポテンシャル)を設定し、ニューラルネットワークに組み込んだ。この関数は、原子間の相互作用・化学結合の強さに対応するため、学習結果の物理化学的な解釈・検証が可能である。
ソースコードを公開中
https://github.com/masashitsubaki/QuantumGNN_molecules
手法の詳細の論文
Masashi Tsubaki and Teruyasu Mizoguchi, Fast and accurate molecular
property prediction: learning atomic Interactions and potentials with neural networks, J. Phys. Chem.
Lett.20189195733-5741, 2018 DOI: 10.1021/acs.jpclett.8b01837
化学物質の物性値を高速・高精度で予測できる本手法によって多数の候補化学物質の物性値を網羅的に評価することで、より優れた機能や新しい機能を持つ化学物質を見出すプロセスを加速することが期待できる。今後は、今回開発した手法をより高精度化するとともに、手法を活用して材料開発を大幅に加速し、新たな化学物質の発見につなげていく。
| 研究開発プロジェクト | NEDO 次世代人工知能・ロボット中核技術開発の成果 |
| 研究機関 | 国立研究開発法人 産業技術総合研究所 |
| 主要研究者 | 椿 真史 |